1. Giải trình tự gen thế hệ mới – Next Generation Sequencing (NGS) là gì?
Vào năm 1977, Fredrick Sanger và các cộng sự đã nghiên cứu và giới thiệu phương pháp kết thúc chuỗi “dideoxy” để giải trình tự các phân tử DNA, hay còn được gọi là “giải trình tự Sanger ”. Từ đó đánh dấu bước ngoặt lớn cho sự phát triển và cải tiến của công nghệ giải trình tự.
Với những tiến bộ công nghệ to lớn này, dự án bộ gen người đã được hoàn thành vào năm 2003. Đến năm 2005, “giải trình tự thế hệ mới NGS”, hay còn gọi là “2G”, đã ra đời. Phương pháp này cho phép khuếch đại hàng triệu bản sao của một đoạn DNA cụ thể dựa trên phương pháp PCR khuếch đại cầu trái ngược với giải trình tự Sanger.
Sự khác biệt chính giữa giải trình tự Sanger và 2G NGS nằm ở khối lượng giải trình tự. Về nguyên tắc của phương pháp giải trình tự thế hệ mới NGS so với giải trình tự Sanger có một số điểm khá tương đồng nhau. Tuy nhiên, hệ thống này lại giải trình tự dài bằng cách phân cắt chúng thành các mảnh nhỏ (các read), có kích thước ngắn khoảng 150-700 bp. Đồng thời NGS còn cho phép xử lý nhiều đoạn read cùng một lúc, dẫn đến tăng năng suất, độ nhạy; tiết kiệm thời gian; cắt giảm chi phí và dữ liệu đầu ra hơn so với Sanger.
Có hai cách tiếp cận chính trong công nghệ NGS
+) Giải trình tự đọc ngắn (short-read)
+) Đọc dài (long-read)
Hiện nay, NGS được ứng dụng trong lâm sàng để chẩn đoán bệnh ung thư, nhiều loại rối loạn di truyền khác nhau thông qua việc xác định các đột biến di truyền (dòng mầm – germline) hoặc đột biến mắc phải (soma). Ngoài ra, NGS cũng là một công cụ có giá trị trong các nghiên cứu metagenomic và được sử dụng để chẩn đoán, theo dõi và kiểm soát các bệnh truyền nhiễm. Vào năm 2020, các phương pháp NGS đóng vai trò then chốt trong việc mô tả đặc điểm bộ gen của SARS-CoV-2 và không ngừng góp phần trong công cuộc phòng chống đại dịch COVID-19
2. Các bước chính của phương pháp giải trình tự gen thế hệ mới – Next Generation Sequencing (NGS)
Bất kể phương pháp 2G NGS được chọn là gì, có một số bước chính phải được điều chỉnh và tối ưu hóa cho mục tiêu (RNA hoặc DNA) và hệ thống giải trình tự đã chọn.
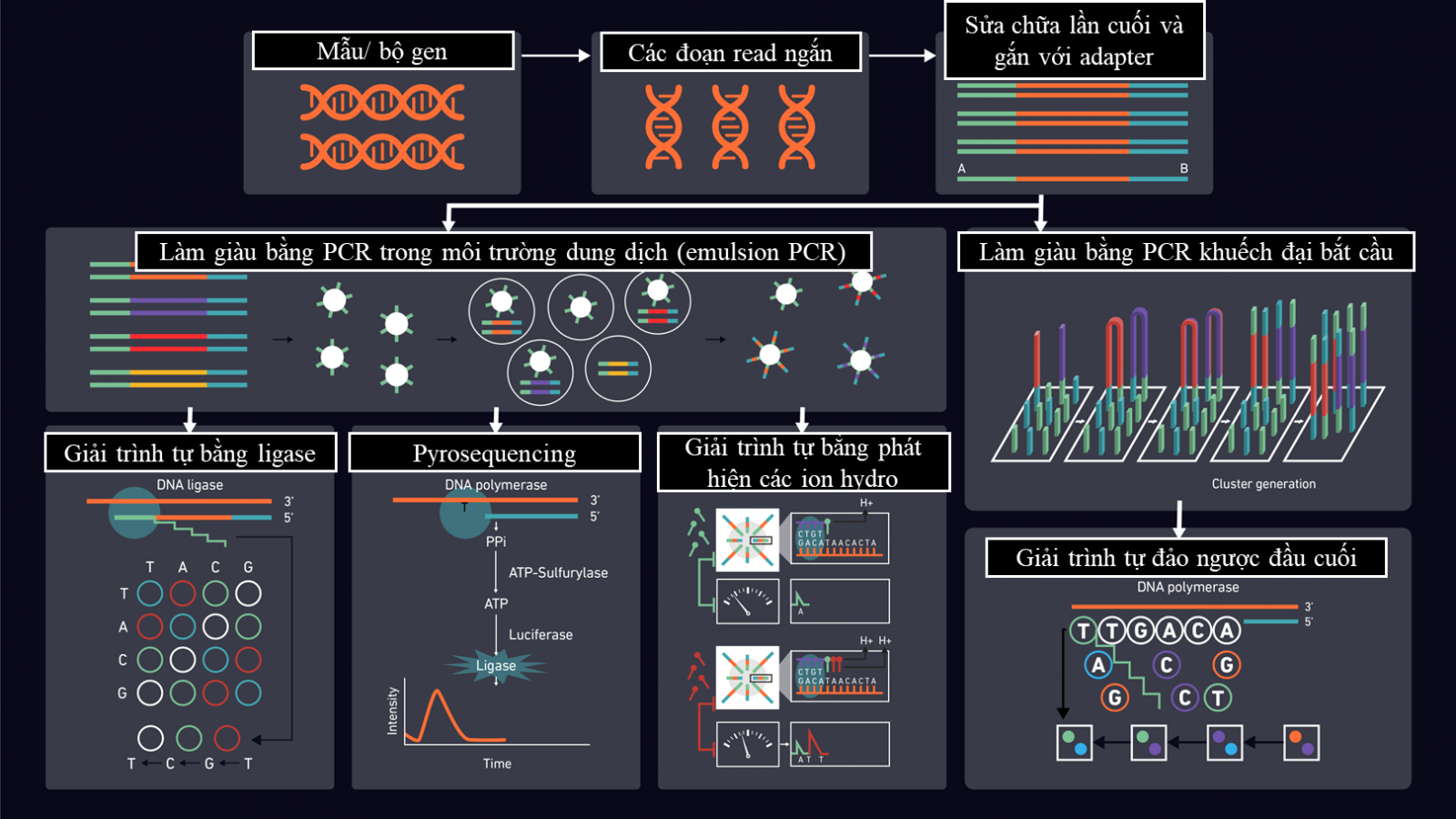
(a) Chuẩn bị mẫu (tiền xử lý)
Nucleic acid (DNA/RNA) được tách chiết từ nhiều loại mẫu khác nhau (máu, đờm, tủy xương,…). Các mẫu được tách chiết sẽ được kiểm tra chất lượng (QC) bằng cách sử dụng các phương pháp tiêu chuẩn (đo quang phổ, đo huỳnh quang hoặc điện di trên gel). Đối với RNA, thì cần phải thực hiện phiên mã ngược thành cDNA, tuy nhiên, một số bộ kit chuẩn bị thư viện có thể bao gồm bước này.
(b) Chuẩn bị thư viện
cDNA hoặc DNA sẽ bị phân mảnh ngẫu nhiên tạo thành các đoạn read ngắn bằng cách sử dụng enzyme hoặc sóng siêu âm. Việc tối ưu hóa độ dài đoạn trình tự phụ thuộc vào dòng máy đang được sử dụng và có thể chạy điện di một lượng nhỏ mẫu chứa các đoạn read khi tiến hành tối ưu hóa quy trình này. Các đoạn read sau đó được sửa chữa lần cuối và nối với các đoạn DNA chung có kích thước ngắn gọi là “Adaptor” ở cuối hai đầu của 3’ và 5’ của các đoạn read. Các Adaptor có độ dài đã được xác định với các trình tự oligome đã biết để tương thích với dòng máy giải trình tự và có thể phân biệt được khi thực hiện giải trình tự ghép kênh.
Giải trình tự ghép kênh, sử dụng các adaptor riêng lẻ trên mỗi mẫu, cho phép gộp một số lượng lớn thư viện và giải trình tự đồng thời trong một lần chạy. Tập hợp các đoạn DNA sau khi gắn adaptor này được gọi là thư viện giải trình tự.
Tiếp theo là bước lựa chọn kích thước phù hợp, bằng điện di trên gel hoặc sử dụng hạt từ tính, để loại bỏ các đoạn trình tự có kích thước quá ngắn hoặc quá dài, điều này sẽ giúp đạt hiệu suất tối ưu hơn trong quá trình giải trình tự.
(c) Làm giàu/khuếch đại thư viện bằng cách phương pháp PCR khuếch đại cầu.
Các đoạn ADN sau khi gắn adapter được tiến hành làm giàu bằng PCR (Polymerase chain reaction) trong môi trường dung dịch (emulsion PCR) để tạo cụm giải trình tự. Quá trình khuếch đại thường được theo sau bởi bước “làm sạch” (ví dụ: sử dụng hạt từ tính) để loại bỏ các đoạn không mong muốn và cải thiện hiệu quả giải trình tự.
Các thư viện cuối cùng có thể trải qua quá trình kiểm tra QC bằng cách sử dụng qPCR để xác nhận chất lượng và số lượng DNA. Điều này cũng sẽ cho phép chuẩn bị đúng nồng độ mẫu để giải trình tự.
Cuối cùng các thư viện có thể trải qua quá trình kiểm tra QC bằng cách sử dụng qPCR để xác nhận chất lượng và số lượng DNA. Điều này cũng sẽ cho phép chuẩn bị đúng nồng độ mẫu để giải trình tự.
(d) Giải trình tự
Tùy thuộc vào dòng máy và hóa chất sử dụng, quá trình khuếch đại vô tính của các doạn thư viện DNA/cDNA có thể được thực hiện trước khi tiến hành sắp xếp giải trình tự. Các trình tự sau đó được phát hiện và ghi nhận lại trên hệ thống.
(e) Phân tích dữ liệu
Các phương pháp phân tích dữ liệu phụ thuộc nhiều vào mục tiêu nghiên cứu. Mặc dù có thể giảm số lượng mẫu mà được phân tích trong một lần chạy nhất định, nhưng giải trình tự paired-end và mate pair mang lại lợi thế trong phân tích dữ liệu xuôi dòng, đặc biệt đối với các tập hợp de novo. Các kỹ thuật liên kết trình tự đọc với nhau được đọc từ cả hai đầu của một đoạn (paired-end) hoặc được phân tách bằng một vùng DNA xen giữa (mate pair).
Rõ ràng có nhiều lựa chọn khi nói đến việc lựa chọn một chiến lược giải trình tự. Sau đây là một số lưu ý khi quyết định lựa chọn phương pháp đọc, sắp xếp trình tự và chuẩn bị thư viện phù hợp:
(a) Câu hỏi nghiên cứu được đặt ra
(b) Loại mẫu
(c) Trình tự đọc ngắn hoặc đọc dài
(d) Giải trình tự DNA hoặc RNA – bạn có cần xem bộ gen hoặc bộ phiên mã không?
(e) Sử dụng toàn bộ bộ gen hay chỉ các vùng trình tự cụ thể?
(f) Độ sâu đọc (độ bao phủ) cần thiết – cụ thể cho thử nghiệm
(g) Phương pháp tách chiết
(h) Nồng độ mẫu
(i) Giải trình tự single end, paired end hay mate pair
(j) Yêu cầu độ dài các đoạn read phải cụ thể, rõ ràng
(K) Các mẫu có thể thực hiện multiplex không?
(l) Các công cụ tin sinh học – phụ thuộc vào thí nghiệm. Tùy thuộc vào mẫu và mục tiêu đã đề ra, toàn bộ quá trình phân tích trình tự có thể được điều chỉnh.
Tài liệu tham khảo
[1] An Overview of Next-Generation Sequencing: http://www.technologynetworks.com/genomics/articles/an-overview-of-next-generation-sequencing-346532. Accessed: 2023-05-29.
[2] Genomics, F.L. and Schroeder, K. (2022), A History of Sequencing. Front Line Genomics.



